The VISION Project The VISION project conducted a ValIdated Systematic IntegratiON of epigenetic datasets across progenitor and differentiated blood cell types in mouse and human (Heuston et al. 2018, Xiang et al. 2020, Xiang et al. 2024). The project was carried out by an international group of scientists funded by the National Institute of Diabetes, Digestive, and Kidney Diseases of the National Institutes of Health (grant R24DK106766) and with intramural support from the National Human Genome Research Institute. Key products and results of the project can be visualized on the UCSC Genome Browser using this track hub. The project website provides other servers, databases, and data downloads.
Candidate cis-regulatory elements, or cCREs A candidate cis-regulatory element is defined as a DNA interval with a high signal for chromatin accessibility in any cell type (Xiang et al. 2020). Almost all well-characterized cis-regulatory elements (CREs) are genomic intervals with high accessibility to nucleases in chromatin, and thus high localized accessibility is used to predict candidate CREs, or cCREs. Such a genomic element is considered a candidate CRE until experimental evidence supports it having a role in gene regulation.
This Candidate CRE super track provides access to three resources:
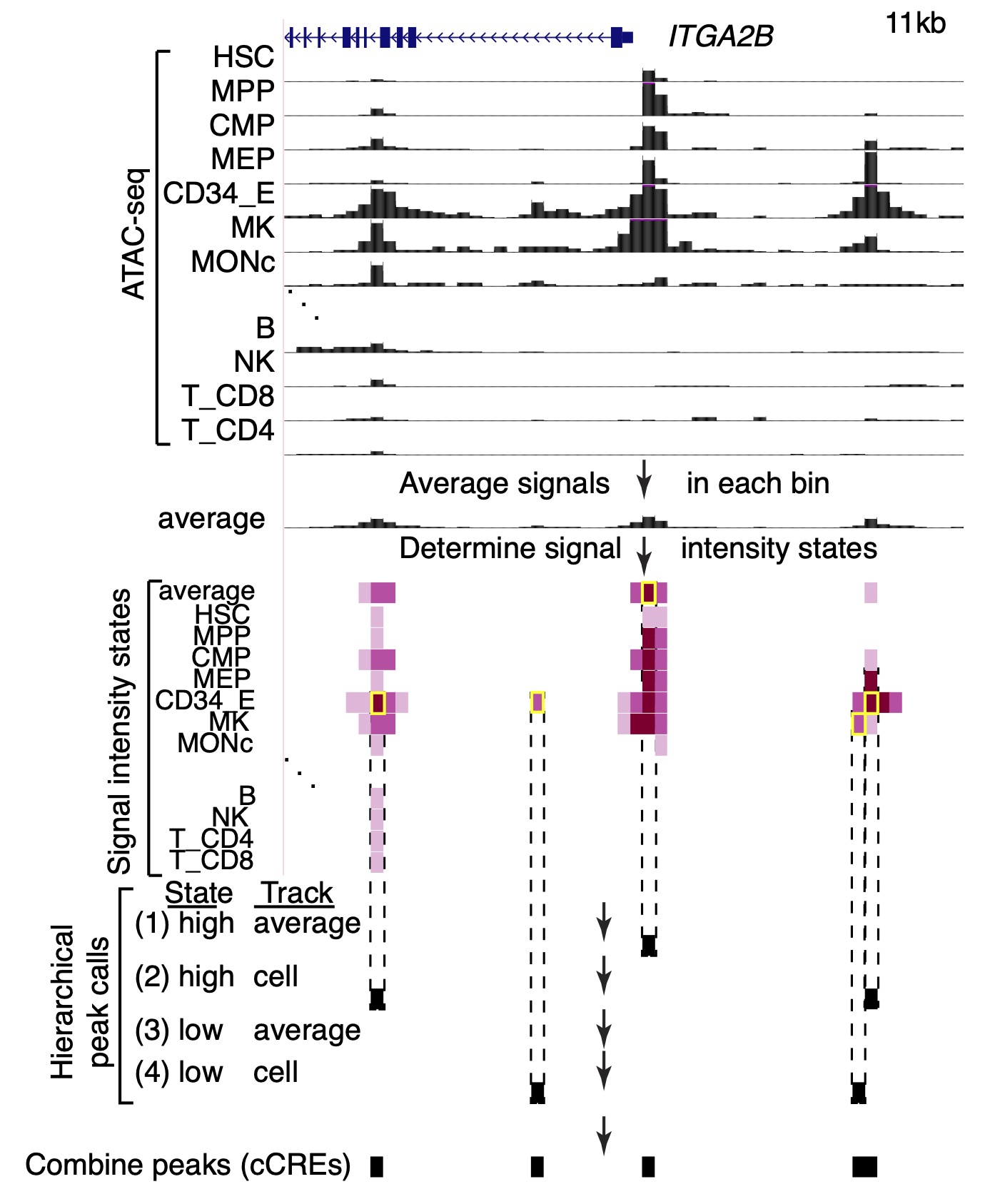
Identifying cCREs from ATAC-seq signal intensity states In order to find localized regions of high accessibility to call as cCREs, the normalized (Xiang et al. 2020 and 2021) ATAC-seq signals across cell types were discretized by using IDEAS (Zhang et al. 2016 and 2017) in the signal intensity state (IS) mode (Xiang et al. 2021 and 2024). In this way, the continuous ATAC-seq signal was converted to four discrete states of signal strength, which were then processed to generate peak calls across cell types, as illustrated in the Figure. This method helps counteract excessive expansion of peak calls when combining them. The resulting peak calls were considered cCREs.
The display conventions are explained on the Track Settings page for each of the component tracks and composite tracks.
The use of IDEAS in the IS mode for peak calling on chromatin accessibility data was described in Xiang et al. 2021 and Xiang et al. 2024. The assignments of cCREs to evolutionary categories and the joint metaclusters were described in Xiang et al. 2024.
Guanjue Xiang performed the data normalization and peak calls using IDEAS in the IS mode. The data downloads, re-mapping and processing, generation of the tracks displayed, and development of the track hub were done by Belinda Giardine.
Heuston EF, Keller CA, Lichtenberg J, Giardine B, Anderson SM; NIH Intramural Sequencing Center; Hardison RC, Bodine DM. Establishment of regulatory elements during erythro-megakaryopoiesis identifies hematopoietic lineage-commitment points. Epigenetics Chromatin. 2018 May 28;11(1):22. PMID: 29807547; PMCID: PMC5971425.
Xiang G, Keller CA, Heuston E, Giardine BM, An L, Wixom AQ, Miller A, Cockburn A, Sauria MEG, Weaver K, Lichtenberg J, Göttgens B, Li Q, Bodine D, Mahony S, Taylor J, Blobel GA, Weiss MJ, Cheng Y, Yue F, Hughes J, Higgs DR, Zhang Y, Hardison RC. An integrative view of the regulatory and transcriptional landscapes in mouse hematopoiesis. Genome Res. 2020 Mar;30(3):472-484. PMID: 32132109; PMCID: PMC7111515.
Xiang G, Keller CA, Giardine B, An L, Li Q, Zhang Y, Hardison RC. S3norm: simultaneous normalization of sequencing depth and signal-to-noise ratio in epigenomic data. Nucleic Acids Res. 2020 May 7;48(8):e43. doi:10.1093/nar/gkaa105. PMID: 32086521; PMCID: PMC7192629.
Xiang G, Giardine BM, Mahony S, Zhang Y, Hardison RC. S3V2-IDEAS: a package for normalizing, denoising and integrating epigenomic datasets across different cell types. Bioinformatics. 2021 Sep 29;37(18):3011-3013. doi:10.1093/bioinformatics/btab148. PMID: 33681991; PMCID: PMC8479670.
Xiang G, He X, Giardine BM, Isaac KJ, Taylor DJ, McCoy RC, Jansen C, Keller CA, Wixom AQ, Cockburn A, Miller A, Qi Q, He Y, Li Y, Lichtenberg J, Heuston EF, Anderson SM, Luan J, Vermunt MW, Yue F, Sauria MEG, Schatz MC, Taylor J, Göttgens B, Hughes JR, Higgs DR, Weiss MJ, Cheng Y, Blobel GA, Bodine DM, Zhang Y, Li Q, Mahony S, Hardison RC. Interspecies regulatory landscapes and elements revealed by novel joint systematic integration of human and mouse blood cell epigenomes. Genome Res. 2024 Aug 20;34(7):1089-1105. PMID: 38951027; PMCID: PMC11368181.
Zhang Y, An L, Yue F, Hardison RC. Jointly characterizing epigenetic dynamics across multiple human cell types. Nucleic Acids Res. 2016 Aug 19;44(14):6721-31. doi: 10.1093/nar/gkw278. Epub 2016 Apr 19. PMID: 27095202; PMCID: PMC5772166.
Zhang Y, Hardison RC. Accurate and reproducible functional maps in 127 human cell types via 2D genome segmentation. Nucleic Acids Res. 2017 Sep 29;45(17):9823-9836. doi: 10.1093/nar/gkx659. PMID: 28973456; PMCID: PMC5622376.
These data are available for use without restrictions.
Ross Hardison rch8@psu.edu